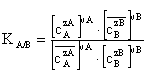
| ↓8 |
Leistungsfähige funktionelle Polymere bestehen aus einer hochmolekularen, organischen Matrix (Grundkörper oder Gerüstbildner), einem Brückenbildner (Vernetzer) und einer austauschaktiven Komponente (Ankergruppe). Die Ankergruppe setzt sich aus Fest- und Gegenion zusammen, wobei nur das Festion (ladungstragende Ankergruppe) kovalent mit dem Grundgerüst verbunden ist. Das Gegenion ist der austauschbare Bestandteil und trägt eine dem Festion entgegengesetzte Ladung. Die heute am häufigsten eingesetzten chelatbildenden Ionenaustauscher haben ein Grundgerüst auf Polystyrenbasis (PS), welches mit Divinylbenzen (DVB) vernetzt wurde und dadurch einen hochmolekularen dreidimensionalen Aufbau aufweist.
Der Ionenaustauschvorgang ist reversibel, stöchiometrisch und elektroneutral [Helfferich 1959]. Ionenaustauscher werden durch Eigenschaften wie Selektivität, Kapazität und Regenerierbarkeit charakterisiert. Für ihre praktischen Anwendungen sind deren Kinetik und die Stabilität entscheidend. Die Einflussfaktoren zur Steuerung der Ionenaustauschereigenschaften lassen sich in drei Gruppen unterteilen: Chemische Zusammensetzung, Adsorptionsbedingungen und Matrixeinflüsse (Abb. 2.1.1). Da diese Faktoren sich gegenseitig beeinflussen ist die Steuerung der Eigenschaften sehr komplex. Die Matrixstruktur beeinflusst die mechanische Stabilität, das Quellvermögen, die Kapazität und die Kinetik des Ionenaustausches. Mit zunehmendem Quellvermögen steigen sowohl die Adsorptionsgeschwindigkeit als auch die Stabilitätskonstanten der Metallkomplexe. Durch gezielte Ligandenauswahl bestimmt man deren Affinität zu ausgewählten Metallionen. Neben der Zusammensetzung und der Struktur der Matrix sind die Austauschereigenschaften von den Adsorptionsbedingungen abhängig. Die Lage des Komplexgleichgewichtes wird von den äußeren Bedingungen, wie pH-Wert, Art der Metallionen, Elektrolytkonzentration und der Temperatur bestimmt [Harland 1994].
| ↓9 |
Durch Variation der experimentellen Bedingungen kann man die Selektivität und Kapazität der Ionenaustauscher gegenüber Metallionen in gewissen Grenzen einstellen.
| Abb. 2.1.1: Einflussparameter auf das Austauschverhalten | ||
|
|
Die Selektivität - Bevorzugung eines Gegenions gegenüber einem anderen - hängt von folgenden Faktoren ab:
| ↓10 |
Die Bindung von Schwermetallionen an IDE-Ionenaustauscher aus neutralen Wässern verläuft entsprechend der nachstehend aufgeführten Selektivitätsreihenfolge [Bayer 2002]:
| ↓11 |
Innerhalb dieser Reihenfolge nimmt der Komplexbildungscharakter der Metalle von links nach rechts ab. Die Selektivität mehrwertiger Ionen wird bei IDE-Austauschern wesentlich durch Aspekte der Koordinationschemie bestimmt, sie nimmt in Richtung fallender Komplexbildungstendenz ab. Die Stabilität der Komplexe korreliert mit dem Ionenradius4, d. h. sie nimmt mit steigendem Ionenradius ab. Links stehende Cu-Ionen bilden sehr stabile Chelatkomplexe. Die dazwischen stehenden Metallionen werden gemäß ihrer Komplexbildungstendenz unter anteiliger Salz- und Komplexbildung gebunden. Bei den Ca- und Mg-Ionen ist der komplexartige Bindungscharakter nur schwach ausgeprägt und die in der Selektivitätsreihe ganz rechts stehenden Alkali-Ionen werden ausschließlich über Ionenbindung gebunden.
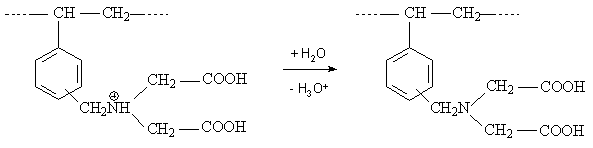
Die Wirkungsweise der funktionellen Gruppen wird im Wesentlichen durch ihr Dissoziationsverhalten bestimmt. Die Lage des Austauschgleichgewichtes ist vom pH-Wert abhängig, da die Carboxylgruppen nicht über den gesamten pH-Bereich in ionischer Form vorliegen. Sie sind im sauren Bereich nur wenig, erst oberhalb vom pH–Wert 6 hinreichend dissoziiert [Kettrup]. Ihr pK-Wert - bei dem 50 % der funktionellen Gruppen in protonierter Form vorliegen - liegt zwischen 4 und 6. Ist der pH-Wert der Lösung kleiner als der pK-Wert des Ionenaustauschers, liegen die Carboxylgruppen weitgehend undissoziiert vor, was einen Ionenaustausch unmöglich macht [Helfferich 1959]. IDE-Ionenaustauscher in der H-Form sind kaum fähig, Metallionen aus saurer oder neutraler Lösung aufzunehmen. Durch die abgegebenen Protonen sinkt der pH-Wert wodurch die Dissoziation der Ionenaustauscher zurückgedrängt wird. In der Praxis werden diese Ionenaustauscher meist in der Alkali- oder Erdalkaliform eingesetzt. Die IDE-Ankergruppen sind in Abhängigkeit vom pH-Wert der Lösung unterschiedlich stark protoniert. Im stark sauren Bereich (pH < 0) lagert sich ein Proton an das Stickstoff-Atom an und es liegt die hydroacide Form vor. Diese hydrolysiert stark und bildet beim Nachwaschen mit Wasser sofort die H-Form (pH 2).
| ↓12 |
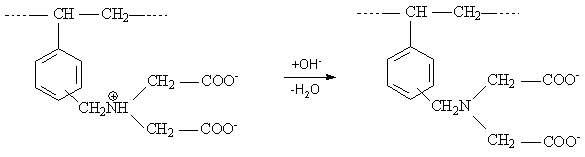
Wird eines der verbliebenen zwei Protonen durch Neutralisation (pH 8) entfernt, wandert das zweite Proton an das Stickstoffatom der funktionellen Gruppe. Durch die Protonenwanderung innerhalb der Ankergruppe bleibt die Symmetrie des Moleküls erhalten und die Donatorwirkung des Stickstoffs wird verstärkt. Bei weiterer Zugabe von Alkalihydroxid (pH 12-13) wird auch das letzte Proton abgelöst und es entsteht die Dialkaliform, welche der Hydrolyse unterliegt und somit stark alkalisch reagiert. Bei andauernder Wasserbehandlung geht die Dialkaliform in die Monoalkaliform über.
Das Vorliegen verschiedener Dissoziationsstufen wird in der Literatur mehrfach durch Aufnahme potentiometrischer Titrationskurven beschrieben [Enders 1990] [Vater 1992]. Chelatbildende Ionenaustauscher haben aufgrund der polaren, hydrophilen funktionellen Carbonylgruppen und des freien Elektronenpaares am Stickstoffatom der IDE-Gruppe eine hohe Affinität zu H+-Ionen, als „Konkurrenz“ zu den Metallionen der Lösung. Die IDE-Gruppe kann als dreizähniger Ligand betrachtet werden. Einerseits sind IDE-Ionenaustauscher durch die beiden Carboxyl-Gruppen zum Kationenaustausch fähig, andererseits können durch das Stickstoffatom Schwermetallionen komplexiert werden, so dass eine zusätzliche Stabilisierung durch den Chelateffekt erreicht wird. In ihrer Struktur entsprechen IDE-Ionenaustauscher einem halben Molekül der EDTA (Ethylendiamintetraessigsäure). Tab. 2.2.1 gibt eine aktuelle Übersicht über kommerziell hergestellte IDE-Ionenaustauscher.
| ↓13 |
Tab. 2.2.1: IDE-Ionenaustauscher verschiedener Hersteller
|
Name |
CAS-Nr.: |
Hersteller/Stadt/Land |
|
Amberlite IRC-718 |
79620-28-3 |
ROHM and HAAS/Philadelphia/USA |
|
Amberlite IRC-748 |
244203-30-3 |
ROHM and HAAS/Philadelphia/USA |
|
Chelex 100 |
11139-85-8 |
BIORAD/Hercules/California |
|
Diaion CR 20 |
57916-98-0 |
MITSUBISHI CHEMICAL INDUSTRIES/ Tokyo/Japan |
|
Dowex A-1 |
9056-04-6 |
DOW CHEMICAL /Midland,Michigan/USA |
|
DUOLITE ES 466 |
73666-75-8 |
ROHM and HAAS/Philadelphia/USA |
|
Imac Syn 101 |
69344-91-8 |
AKZO-CHEMIE/Amsterdam/Niederlande |
|
Ionac SR-5 |
141489-98-7 |
SYBRON CHEMICALS/Birmingham/USA |
|
Ligandex I |
82497-10-7 |
REANAL/Budapest/Ungarn |
|
Lewatit TP 207 |
57285-14-0 |
BAYER AG/Leverkusen/Deutschland |
|
Lewatit TP 208 |
126602-47-9 |
BAYER AG/Leverkusen/Deutschland |
|
Purolite S-930 |
186397-72-8 |
PUROLITE INTERNATIONAL/ Philadelphia/USA |
|
Unicellex, UR 10 |
63590-14-7 |
Unitika/Tokyo/Japan |
|
Varion CH |
58206-59-0 |
Nitrokemia/Veszprem/Ungarn |
Die Einsatzmöglichkeiten chelatbildender Ionenaustauscher sind breit gefächert. Im technischen Maßstab werden die Selektivaustauscher häufig bei der Aufbereitung von Ab- und Prozesswässern der metallver- und -bearbeitenden Industrie eingesetzt. Dort werden die Schwermetallkonzentrationen von einigen mg/L auf unter 1 mg/L gesenkt [Calmon 1981]. Beispielhaft sind hier die
| ↓14 |
Bei der Aufbereitung von Trinkwässern erfolgte der Ionenaustauschereinsatz vorwiegend zur Wasserenthärtung, -entsalzung und zur Herstellung von ultrareinem Wasser [Höll, Flemming 1991]. Erst im letzten Jahrzehnt wurden zahlreiche Untersuchungen zum Einsatz von IDE-Austauschern zur Schwermetallentfernung in der Trinkwasseraufbereitung durchgeführt. Es wurden vorwiegend Ni-Ionen selektiv gegen Ca-Ionen ausgetauscht und aus dem Trinkwasser – bei Erhalt der Wasserqualität - entfernt [Rahm 1994], [Rahm, Overath 1995], [Overath et al. 2002], [Stetter et al. 2006].
Ionenaustauscher mit IDE-Ankergruppen sind nicht nur zur ionischen Bindung von Kationen über die Ladungen der Carboxylgruppen in der Lage, sondern zusätzlich zur Komplexierung zwei- und dreiwertiger Übergangsmetallionen fähig. Komplexbildende Eigenschaften zeigen insbesondere Ionen mit Elektronen in den d- und f-Orbitalen.
Ionen mit Elektronen nur in den s- und p-Orbitalen besitzen eine geringe Neigung zur Ausbildung von Komplexen. Deshalb kommt mit Alkali- und Ammoniumionen keine Komplexbildung zustande. Sie werden nur ionogen über eine Ionenpaarbindung gebunden. Ihr Anteil ist pH-abhängig und ein ausreichender Ionenaustausch erfolgt – bedingt durch die geringe Dissoziation der funktionellen Ankergruppen – erst bei pH-Werten > 7 (vgl. 2.2). Die Stärke der Coulomb’schen Kräfte ist im Wesentlichen von der Ionenladung und den Ionenradien (kürzester Abstand zwischen Festion und solvatisiertem Gegenion) abhängig. Bei der Reaktion mit zwei- und dreiwertigen Metallionen werden zwei Grenzfälle unterschieden: Die ionogene und koordinative Bindung.
| ↓15 |
Bei der koordinativen Bindung werden die Metallionen über eine Donor-Akzeptor-Wechselwirkung an die im Polymer befindlichen funktionellen Gruppen gebunden. Hierbei stellen das Metallion die LEWIS-Säure (Elektronenpaarakzeptor) und die Komplexliganden die LEWIS-Basen – die allein die Bindungselektronen für die koordinative Bindung zur Verfügung stellen – dar. Besitzt ein Ligand mehrere Atome mit freien Elektronenpaaren, so kann dieses Molekül mehrfach an das Zentralatom gebunden sein. Von den Liganden mit anorganischen Donorgruppen sind diejenigen mit O-, S-, N- oder P-Donorgruppen die bedeutendsten, deren freie Elektronenpaare die Koordinationsstellen für das Kation bilden. Die Bindungsart spielt dabei keine Rolle [Lindner 1993]. Es entstehen ringförmige mehrzähnige Chelatkomplexe, z. B. Oxalat, Tartrat, Ethylendiamintetraacetat (EDTA), die eine definierte räumliche Anordnung mit dem Ziel bilden, eine geringe Abstoßung der Liganden untereinander und die größtmögliche Anziehung durch das Metallion zu gewährleisten. Die geometrische Anordnung und die Bindungslängen (Abstand) der Donatoratome des Liganden werden durch das Metallion bestimmt und ergeben für jedes Metallion spezifische Werte.
In Polymerkomplexen werden, aufgrund der eingeschränkten räumlichen Flexibilität der makromolekularen Liganden, die Metallionen meist mit den Koordinationszahlen (KZ) 4 (tetraedrisch, quadratisch-planar) und 6 (oktaedrisch) belegt. Die chelatbildenden IDE-Ankergruppen besetzen aber bei der Komplexbildung stets weniger als 6 Koordinationsstellen des Zentralatoms. Vakante Bindungsstellen werden nicht selten von Lösungsmittelmolekülen oder anderen in der Lösung enthaltenen Substanzen eingenommen. Der Grund hierfür liegt in der gegenseitigen sterischen Behinderung benachbarter funktioneller Gruppen im Makromolekül [Stetter 2004].
Die Bindung eines Liganden an ein Metallkation ändert die Eigenschaften des Liganden drastisch. Es kommt zu Konformationsänderungen des Ligandenmoleküls als Folge der Beteiligung des Liganden -Elektronenpaares an der Bindung zum Metall. Die Umwandlung eines freien zum bindenden Elektronenpaar führt zu Änderungen der Bindungslängen und Bindungswinkel. Zusätzlich übt das Metallion elektrostatische Wechselwirkungen auf das bindende Elektronenpaar des Liganden aus.
| ↓16 |
Chelatkomplexe besitzen meist hohe Komplexbildungskonstanten und haben sowohl thermodynamisch als auch kinetisch eine höhere Stabilität als Komplexe mit einzähnigen Liganden. Das System strebt ein Minimum der freien Enthalpie an, welches auf einem Anstieg der Entropie zurückgeht (Gibbs-Helmholtz-Gleichung). Die thermodynamische Stabilität beruht auf der Erhöhung des Entropieterms des Systems, da bei der Bildung von Chelatkomplexen mehr Teilchen freigesetzt werden als vor der Reaktion vorhanden waren. Das kann man anschaulich mit der Erhöhung der Teilchenzahlen – aufgrund der stufenweisen Substitution von Wassermolekülen – durch Komplexbildung erklären. Bei der Anlagerung eines Chelat-Liganden werden mehrere Wassermoleküle aus dem Aquakomplex verdrängt. Die Zahl der frei beweglichen Teilchen und damit der Entropieterm nehmen zu. Die kinetische Stabilität beruht darauf, dass bei gleicher Konzentration der Liganden die Wahrscheinlichkeit (Geschwindigkeit) zur Bildung der ersten Donor-Zentralatom-Wechselwirkung nahezu gleich groß ist. Für die zweite Koordinationsstelle ist die Wahrscheinlichkeit der Zweitsubstitution für den Chelatliganden wegen der räumlichen Nähe des zweiten Donoratoms größer.
Kennzeichnend für Übergangsmetalle sind partiell gefüllte d-Orbitale – Hauptursache für ihre große Neigung zur Komplexbildung. Jedes Metall besitzt eine mit der Anzahl der d-Elektronen in Beziehung stehende bevorzugte Koordinationszahl und -geometrie. Metallkationen der 3 d-Übergangsmetalle sind meist von sechs Wassermolekülen umgeben. Sie bilden oft farbige Hexaaquakomplexe wie z. B. [Ni(OH2)6]2+ [Rauscher et al. 1976]. Eine Auswahl physikalischer Daten für die im Rahmen dieser Arbeit relevanten Metallionen ist in Tab. 2.3.1 zusammengestellt. Die Bindung in Komplexen von Nebengruppenkationen, welche sich aus elektrostatischen und kovalenten Anteilen zusammensetzen, ist abhängig von der jeweiligen d-Elektronenkonfigurationen und der Größe des Kations sowie vom Liganden mit der Art, Anzahl und Anordnung seiner Donoratome [Langer 2005].
Komplexe sind besonders stabil, wenn das Metallion durch koordinative Bindung die Elektronenkonfiguration des folgenden Edelgases (Krypton 4s24p6, Xenon 5s25p6 bzw. Radon 6s26p6) erreicht. Im Austauscher führt Komplexbildung zu Spannungen (Deformationen), die nicht alle Anordnungen zulassen und somit die Selektivität des Austauschers beeinflussen [Vater 1992].
| ↓17 |
Tab. 2.3.1: Vergleich wichtiger Eigenschaften der untersuchten Metallionen
|
Metallion |
Oz |
Periode |
Ionenradien [nm] |
Hydratations- |
1. Ionisierungs-energie [eV] |
Endkonfiguration |
|
Ca2+ |
20 |
4 |
0,106 |
0,6 |
6,1 |
3s23p6 |
|
Co2+ |
27 |
4 |
0,082 |
* |
7,9 |
3s23p63d7 |
|
Ni2+ |
28 |
4 |
0,078 |
* |
7,6 |
3s23p63d8 |
|
Cu2+ |
29 |
4 |
0,072 |
0,6 |
7,7 |
3s23p63d9 |
|
Zn2+ |
30 |
4 |
0,083 |
0,6 |
9,4 |
3s23p6 3d 10 |
|
Cd2+ |
48 |
5 |
0,103 |
0,5/0,426 |
9,0 |
4s24p6 4d 10 |
|
Pb2+ |
82 |
6 |
0,132 |
0,45/0,401 |
6,1 |
5d 10 6s2 |
In Anlehnung an die HSAB (Hard and Soft Acids and Bases) von Pearson [Pearson 1968] sind nur die Cd-Ionen als weiche Säure einzuordnen, die mit den als hart geltenden IDE-Liganden nur schwache Komplexe bilden. Die übrigen Metalle stellen einen Übergangsbereich dar, weshalb man sie als „Borderline“ - Ionen bezeichnet.
Interessant ist die Pseudoedelgaskonfiguration der Zn- und Cd-Ionen. Metallkationen mit voll besetzten d-Orbitalen wie Zn2+ (3d10), Cd2+ (4d10), Pb2+ (5d10s2) - auch B-Kationen genannt - stellen so genannte „Grenzfälle“ im Sinne des HSAB-Konzeptes dar, da ihre d-Orbitale für eine Bindung nicht zur Verfügung stehen. Bei ihnen spielen Ladung und Radius keine wesentliche Rolle, sie sind relativ leicht deformierbar und bilden kovalente Bindungen aus [LB 5]. Ein geringer Unterschied in den Elektronegativitäten der Bindungspartner wird zum dominierenden Faktor. Die Komplexbildungstendenz nimmt mit zunehmender Elektronegativität der Liganden ab.
| ↓18 |
Metallkationen, deren Elektronenhülle keinen Einfluss auf die Struktur des Komplexes haben, bilden mit vier Liganden meist eine tetraedrische bzw. quadratisch-planare Koordination, die auf eine (sp3) - Hybridisierung der Bindungselektronen am Koordinationszentrum zurückzuführen ist. Das gilt für Ionen mit Pseudoedelgaskonfiguration und Übergangsmetallionen, die andere Anordnungen nicht begünstigen (Co2+). Die KZ 6 ist bei den Cd-Komplexen mit Abstand die häufigste; als zweite typische KZ ist 4 zu nennen. Regulär oktaedrische bzw. tetraedrische Anordnungen werden dabei selten gebildet. Das Cd-Ion (weiche LEWIS-Säure, hohe Polarisierbarkeit) neigt in seinen Komplexen zu verzerrten Geometrien, da es als (d10) - Kation keine zusätzliche Ligandenfeldstabilisierung erfährt
[Johanning 1999].
Wesentliche Einflussgrößen der Komplexbildung der Übergangsmetallkationen mit teilweise besetzten d-Orbitalen wie Co2+(d7), Ni2+(d8), Cu2+(d9) sind Ladung, Größe und Ionisierungsenergie. Sie zeigen eine - durch die d-Orbitalaufspaltung im Ligandenfeld bedingte – Ligandenfeldstabilisierungsenergie (LFSE), welche bei Kationen mit vollbesetzten d-Orbitalen (z. B. Zn2+) nicht auftritt. Bei elektrostatischer Bindung sind Komplexe mit Ionen höherer Wertigkeit stabiler. Die kovalente Bindungsart (Bindungselektronen liefern Liganden) wird durch niedrige Ionenladung und große Ionenradien begünstigt. Während der Ionenradius die elektrostatische Komponente bestimmt, ist die Ionisierungsenergie ein Maß für den erzielten Energiegewinn bei der Komplexbildung durch das Donorelektronenpaar.
Die (d9)-Systeme (Cu2+ mit einem ungepaarten Elektron) bevorzugen in Komplexen die Koordinationszahlen 4 und 6 unter Ausbildung quadratisch-planarer und oktaedrischer Koordinationsgeometrien. Beim oktaedrischen Ligandenfeld sind die Oktaeder aufgrund des auftretenden Jahn-Teller-Effektes7 verzerrt und in z-Richtung gedreht. Dies führt zu einer energetisch günstigeren Orbitalstellung. Ganz allgemein hat die Ligandtopologie entscheidenden Einfluss auf die Komplexbildung mit Metallkationen. Wichtig ist, die Wechselwirkungen zwischen der polymeren Matrix und den Ankergruppen nicht zu vernachlässigen. So üben der Benzylrest, die PS-Kette, der Vernetzer und die unbesetzten Benzenkerne zusätzliche Einflüsse auf die funktionellen Gruppen aus.
| ↓19 |
Da die Komplexbildung vom Größenverhältnis Ligand/Kation abhängig ist, spielen neben der Ligandtopologie die Größe der zu komplexierenden Kationen sowie die möglichen Donorstellen eine wichtige Rolle [Wichmann 2003]. Kleinere Ionen bevorzugen die Koordinationszahl 6 und bilden oktaedrische Koordinationspolyeder.
Liegt eine reine koordinativeBindung vor, so bilden diese zweiwertigen Metallionen im schwach sauren und neutralen Bereich unter Protonenabgabe mit Iminodiessigsäure ausschließlich 1:1 Komplexe, die durch ihre typische Ringstruktur gekennzeichnet und durch Säuren wieder zerlegbar sind. Bei der Säureelution im Säulenverfahren erscheint das jeweilige Metallion mit genau reproduzierbarem Dekomplexierungs-pH-Wert (DpH).
| ↓20 |
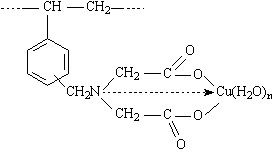
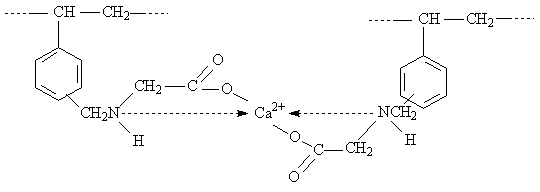
Die IDE-Ankergruppe kann als dreizähniger Ligand betrachtet werden, wobei durch den Chelateffekt eine zusätzliche Stabilisierung der Bindung erreicht wird. Bei der 1:1 Komplexbildung werden drei Koordinationsstellen des Zentralatoms besetzt. Zur Erreichung einer bevorzugten Koordinationsgeometrie können die freien Koordinationsstellen durch bewegliche Fremdliganden komplettiert werden, z. B. durch zusätzliches Wasser, wie es das obige Beispiel zeigt. Zwischen den Wassermolekülen und den Liganden wirken elektrostatische Kräfte unterschiedlicher Stärke und Reichweite in Form von Ion-Dipol- und Dipol-Dipol-Wechselwirkungen. Zusätzlich müssen die Koordination an die d-Metallionen und die Ausbildung von H-Brücken zu den Elektronenpaardonoren beachtet werden.
Zweiwertige Schwermetalle bilden mit IDE-Ionenaustauschern im schwach sauren und neutralen Bereich ausschließlich 1:1 Komplexe. Sie erfahren zwar mit steigendem pH-Wert eine Stabilitätsschwächung, doch kommt es aufgrund sterischer Behinderungen nicht zur Ausbildung von
1:2 Komplexen. Die zweite IDE-Ankergruppe kann dem 1:1 Komplex nur unter Deformation (Energiezufuhr) genähert werden. Erst bei hohen pH-Werten (über 8…9) bilden sich doppelt verankerte
1:2 Komplexe, die wegen des sterischen Zwanges (Spannungscharakter vielgliedriger Ringe) relativ instabil sind [Hering 1967].
Die hohe Selektivität der IDE-Ionenaustauscher für Schwermetalle beruht darauf, dass die anderen in natürlichen Wässern vorkommenden Kationen (Alkali- und Erdalkali-Ionen) keine oder nur sehr schwache Elektronenpaarakzeptoren sind und keine oder nur Komplexe mit sehr geringer Stabilität bilden [Höll 2001]. Werden die zweiwertigen Metallionen vorwiegend ionogen gebunden, so ergibt sich ein molares Verhältnis von 1:2 (Me2+/IDE-Gruppen), d. h. nur die gleichzeitige Beteiligung zweier verankerter Liganden führt zu einer stabilen Verbindung.
| ↓21 |
Diese Bindung liegt vorwiegend bei Erdalkali-Ionen vor und ist weniger stabil als 1:1 Komplexe. Sie ist nur dann existenzfähig, wenn die bei der Komplexbildung frei werdende Komplexbildungsenergie größer der Deformationsenergie ist, welche aufgewendet werden muss, um Ankergruppen und Ionenaustauschermatrix in die geometrische Anordnung zu bringen. Die Deformationsenergien werden mit zunehmendem Volumen des Zentralatoms kleiner [Hering 1967].
Bei der Bindung von zweiwertigen Metallionen an IDE-Gruppen sind mehr oder weniger beide Bindungsformen vorhanden. Der Anteil der jeweiligen Bindungsart hängt von der Komplexbildungstendenz der Metallionen ab. Die Bindung kann prinzipiell intramolekular oder auch intermolekular durch zwei Polymerstränge erfolgen. Die Beteiligung von mehr als zwei Polymermolekülen an der Komplexierung ist aufgrund des sterischen Einflusses des Polymerrückgrats eher unwahrscheinlich. Je niedriger die Stabilität der gebildeten Komplexe, desto größer ist der ionogene Bindungsanteil (Salzcharakter).
| ↓22 |
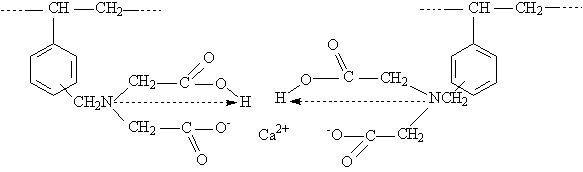
Jede Aminosäureankergruppe kann als zweizähniger Ligand angesehen werden, sofern sie dissoziiert vorliegt. Sie besetzt dann bei der 1:2 Komplexbildung vier Koordinationsstellen (N-Atom der Aminogruppe für die koordinative Bindung und O-Atom der Carboxylgruppe für die ionische Bindung) und zeigt eine tetraedrische bzw. quadratisch-planare Koordination. Komplexe an Polymeren mit Aminosäureankergruppen sind wesentlich instabiler als an IDE-Ankergruppen. So liegt z. B. der DpH des Kupfers bei 2,4 (vgl. IDE bei 1,25) [Hering 1967].
Diese weniger stabilen Komplexe bilden sich vorwiegend bei einer Probe mit niedrigem Zweitsubstitutionsgrad – Probe 1 (TK/N 1,034) – bei der nur 51,7 % der Ankergruppen funktionalisiert sind. Die funktionellen Gruppen liegen hier zu einem sehr großen Anteil als Aminoessigsäure vor.
| ↓23 |
Als Maß für die Komplexstabilität sind die Stabilitätskonstanten (ßn) der Komplexbildungsreaktionen gemäß der allgemeinen Gleichungen 2.1 und 2.2 für wässrige Lösungen in Standardwerken tabelliert. Die Konstanten werden mit zunehmender Zahl der Liganden infolge von statischen, elektrostatischen und sterischen Wechselwirkungen kleiner.
|
(Gl. 2.1) |
|
|
(Gl. 2.2) |
Die Stabilität der Komplexe steigt im Allgemeinen mit dem pH-Wert, was auf die stärkere Dissoziation der funktionellen Gruppen zurückzuführen ist. Der Anstieg der Komplexstabilität mit steigendem pH-Wert wird durch die Ausfällung von Metallhydroxiden begrenzt [Schinner, Sonnleitner 1997].
| ↓24 |
Die Trennung der Metallionen geht um so leichter, je stärker sich die betreffenden Kationen im Selektivitätsverhalten unterscheiden. Die Irving-Williams-Reihe [Irving, Williams 1948] stellt eine gute Möglichkeit dar, die Komplexstabilitäten zweiwertiger Ionen der ersten Übergangsmetallreihe abzuschätzen.
Ba2+ < Sr2+ < Ca2+ < Mg 2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+
Sie beschreibt die Tendenzen für Reaktionen aus Hexaaquakomplexen zweiwertiger Ionen in high-spin-Konfiguration. Als Gründe für das Auftreten dieser Reihenfolge ist neben dem abnehmenden Ionenradius der Beitrag der Orbitalstabilisierungsenergie von entscheidender Bedeutung. Er steht für den Beitrag, der bei der Komplexbildung des oktaedrischen Komplexes gewonnen wird und ist abhängig von der Ligandenfeldaufspaltung [Sigel, Mc Cormick 1970].
| ↓25 |
Bedingt durch die funktionellen Gruppen zeigen chelatbildende Ionenaustauscher zusätzlich zu den ionogenen und koordinativen Bindungen für Kationen auch Anionenaustauschereigenschaften, speziell gegenüber Anionen sehr starker Säuren (ClO4 -, I-, IO3 -; IO4 -, CrO4 2- ). In der Praxis spielen die Chlorokomplexe eine besondere Rolle [Hering 1967].
Der Dekomplexierungs-pH-Wert (DpH) ist der pH-Wert, bei dem das betreffende Metallion gerade wieder vom Ionenaustauscher desorbiert wird. Aufgrund der unterschiedlichen Komplexbildungstendenzen der einzelnen Metalle ist der DpH eine charakteristische und reproduzierbare Größe für jedes Metall und darf beim Beladungsvorgang nicht unterschritten werden. Das Maximum der Aufnahmekapazität wird für ein bestimmtes Metall erst ab einem Zulauf-pH-Wert von 2 Einheiten über dem jeweiligen DpH und höher erreicht. Bei einem Zulauf-pH-Wert von ca. 0,5 – 1 Einheiten über dem jeweiligen DpH ist nur eine eingeschränkte Aufnahme möglich. Die Kenntnis der DpH-Werte ist für die Trennung von Metallgemischen von praktischer Bedeutung (Tab. 2.4.1).
Tab. 2.4.1: Dekomplexierungs-pH-Werte ausgewählter Metallionen [Bayer 2002]
|
Element |
Cu2+ |
Pb2+ |
Ni2+ |
Zn2+ |
Co2+ |
Cd2+ |
Ca2+ |
|
DpH-Wert |
1,00 |
1,80 |
2,10 |
2,50 |
2,55 |
2,80 |
4,40 |
| ↓26 |
Je nach Bedarf müssen zur selektiven Bindung eines Großteils der umweltrelevanten Metallionen die IDE-Ionenaustauscher vor Gebrauch mit Alkali-, Erdalkali- oder Ammoniumionen vorbeladen, d. h. konditioniert werden.
Die jeweilige Lieferform (meist Di-Na-Form) wird zuerst mit Säure regeneriert und somit in die Wasserstoff-Form (H-Form) überführt. Die Anwendung der reinen H-Form beschränkt sich vorwiegend auf die Bindung von Kupferionen. Sie ist Ausgangspunkt für alle weiteren Einsatzformen.
Eine partiell beladene Mono-Na-Form (50 % der Totalkapazität (TK) mit Na+-Ionen belegt) wird vorzugsweise bei Ca2+/Mg2+-armen Zulauflösungen, z. B. bei NaOH-neutralisierten Abwässern eingesetzt.
| ↓27 |
Die Di-Na-Form (100 % der TK mit Na+-Ionen belegt) wird zur Reinigung kalkneutralisierter Abwässer verwendet. Bereits beim Waschen des Ionenaustauschers mit Wasser hydrolysiert die Di-Natrium-Form. Der Filterablauf ist zu Beginn der Versuchslaufzeit aufgrund der hydrolysierenden Natrium-Ionen stark alkalisch (pH>10), so dass ein Waschvorgang bis zur Alkalifreiheit des Ablaufes problematisch ist. Sie wird deshalb meist verwendet, wenn bei einem pH-Wert über 8,5 gearbeitet werden soll.
Oft enthalten in der Praxis zu behandelnde Wässer bzw. Abwässer eine Vielzahl von Salzen, deren Ionen am Ionenaustauscher mit um die verfügbaren Austauschplätze konkurrieren. Deshalb wird für kalkneutralisierte Abwässer sowie bei der Grundwassersanierung ebenfalls die Ca-Form (100 % der TK mit Ca2+-Ionen belegt) eingesetzt. Der Filterablauf ist im gesamten Laufzeitbereich neutral.
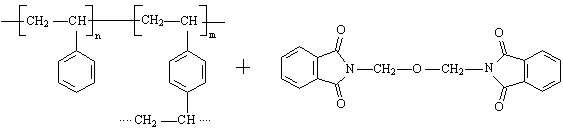
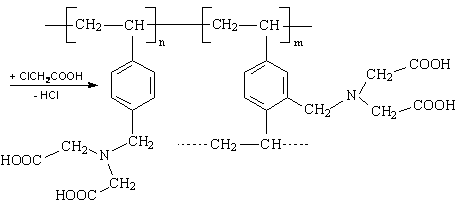
Für die Synthese von chelatbildenden IDE-Ionenaustauschern sind aus der Literatur mehrere Synthesewege bekannt, die alle als Matrix ein inertes, vernetztes Polystyren-Grundgerüst enthalten. Zur Funktionalisierung wird in der Literatur meist die Chlormethylierung mittels Chlordimethylether beschrieben. Nach Pepper und Hale werden durch Aminierung mit Ammoniak und anschließender Carboxymethylierung der Aminogruppen mit Chloressigsäure die funktionellen IDE-Gruppen eingefügt [Pepper, Hale 1954].
| ↓28 |
Einen ausführlichen Überblick hierzu gibt Hering in seinem Standardwerk [Hering 1967].
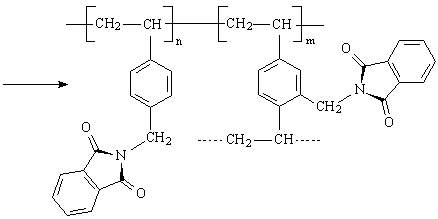
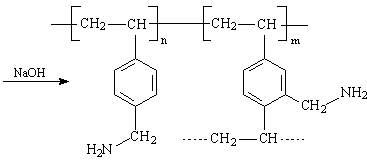
Im Rahmen dieser Arbeit wurden monodisperse Perlpolymerisate auf Styren-Divinylbenzen Basis mit einer Vernetzung von 6 % Divinylbenzen (DVB) eingesetzt. Dieses Polymerisat wurde mit Phthalimidderivaten amidomethyliert und mit Natronlauge zu aminomethyliertem Perlpolymerisat verseift.
| ↓29 |
Vorteil dieser Synthese ist, dass die durch Hering für die Chlormethylierung beschriebenen Quervernetzungen und Nebenankergruppen nicht auftreten.
| ↓30 |
Es erfolgte eine Substitution von H-Atomen der aromatischen Kerne der im Polymeren eingebauten Styren- und Divinylbenzenmonomeren, wobei maximal 5 H-Atome am Kern des Styren und 4 H-Atome am Kern des Divinylbenzen einer unendlichen Zahl von Monomeren zur Verfügung stehen. Die bereits am Phenylring befindlichen Substituenten dirigieren die Substitution der H-Atome durch die Aminomethyl-Gruppen in p- und o-Stellung. Durch IR-Untersuchungen wurde festgestellt, dass die o-Position jedoch selten auftritt. Der aromatische Benzenkern setzt durch Elektronenentzug die Basizität des Stickstoffs herab und fördert damit die Ablösung und Aminierung.
Dieses aminomethylierte Perlpolymerisat diente als Ausgangsstoff zur Herstellung unterschiedlich substituierter Proben. Damit haben alle Proben eine einheitliche Erstsubstitution. Die Charakterisierung dieses Eduktes wurde durch CHN–Elementaranalyse vorgenommen. Im Mittel wurde ein Substitutionsgrad von 1,15 erreicht, d. h. es wurden 1,15 H-Atome durch CH2-NH2-Gruppen ersetzt. Durch stufenweise Umsetzung mit Chloressigsäure wurden die chelatbildenden Ankergruppen erzeugt. Für die experimentellen Untersuchungen wurden 6 Labormuster vom Typ TP 207 mit differenziertem Substitutionsgrad hergestellt. Ausgehend von einer konstanten Menge an aminomethyliertem Perlpolymerisat (einheitlicher Erstsubstitution) wurde die Einsatzmenge an Chloressigsäure schrittweise erhöht. Aus dem Verhältnis der Totalkapazität (TK) und dem Gesamtstickstoffgehalt (N) lassen sich Aussagen über den Substitutionsgrad machen. Durch Wahl der Synthesebedingungen wurde das TK/N-Verhältnis zwischen 1 und 2 variiert.
| ↓31 |
Ein TK/N-Verhältnis von 1 bedeutet, dass nur ein Proton der NH2-Gruppe gegen eine funktionelle Gruppe substituiert wurde und Aminoessigsäure vorliegt. Beim TK/N-Verhältnis 2 sind beide Protonen ausgetauscht und es entsteht Iminodiessigsäure.
Im Rahmen dieser Arbeit wurde als Vergleichssubstanz der käuflich erworbene Ionenaustauscher
TP 207 eingesetzt. Lewatit TP 207, CAS-Nr. 57285-14-0, ist ein schwach saurer, makroporöser, metallspezifischer Ionenaustauscher der Bayer AG Leverkusen8. Dieser wird auf Basis von Styren-Divinylbenzen-Copolymerisaten mit einer Vernetzung von 8 % DVB hergestellt und enthält Iminodiessigsäure (IDE) als funktionelle Gruppen. Die Auslieferung erfolgt in der Di-Na-Form. Die Produktdaten des Herstellers des verwendeten TP 207 sind in Tab. 2.7.1 dokumentiert.
| ↓32 |
Vor Versuchsbeginn wurde das Vergleichsmaterial zunächst einem mehrmaligen Beladungswechsel mit H- und Na-Ionen unterzogen, um die durch den Herstellungsprozess entstandenen Verunreinigungen zu entfernen. Vor dem ersten Beladungszyklus erfolgte die Konditionierung gemäß Kapitel 3.1.2 in die Ca-Form.
Tab. 2.7.1: Produktdaten Lewatit TP 207 [Bayer 2002]
|
Hersteller |
Bayer AG Leverkusen |
|
Probetyp |
Schwach saurer, chelatbildender Kationenaustauscher |
|
Matrix |
Vernetztes, makroporöses Polystyren |
|
Funktionelle Gruppe |
Iminodiessigsäure |
|
Lieferform |
Dinatriumform |
|
Dichte [g/cm³] |
1,17 |
|
Korngrößenbereich > 90 % [mm] |
0,4 – 1,25 |
|
Effektive Korngröße [mm] |
0,55 (± 0,05) |
|
Wassergehalt [%] |
53 – 58 |
|
pH-Arbeitsbereich |
1,5 – 9 |
|
Totale Kapazität [eq/L] |
2,2 (H-Form) |
|
Volumenänderung Na+ →H+ max. [%] |
- 35 |
|
Beständigkeit im pH-Bereich |
0 - 14 |
|
Regeneration |
HCl, H2SO4, HNO3 |
Die Schwermetalle werden in Form ihrer Kationen - vorzugsweise im schwach sauren bis schwach alkalischen Milieu - vom Ionenaustauscher TP 207 gebunden. Die chemischen Eigenschaften der Ankergruppen und der Gegenionen haben zur Folge, dass die verschiedenen Ionen unterschiedlich gut sorbiert werden. Die Bindung der Schwermetall-Ionen aus neutralen Wässern verläuft entsprechend der nachstehend aufgeführten Selektivitätsreihenfolge [Bayer 2002]:
| ↓33 |
Kupfer > Vanadium > Uran > Blei > Nickel > Zink > Kobalt > Cadmium > Eisen(II) > Beryllium > Mangan > Calcium > Magnesium > Strontium > Barium > Natrium
Überlässt man eine beliebige Ionenaustauschreaktion sich selbst, so stellt sich nach einer bestimmten Zeit ein Gleichgewichtszustand (GGW) ein.
|
(Gl. 2.3) |
| ↓34 |
(Überstrichene Größen sind austauschaktiver Bestandteil am Ionenaustauscher)
In Kontakt mit einer Elektrolytlösung, die andere Gegenionen als der Austauscher selbst enthält, gibt der Ionenaustauscher einen Teil seiner ursprünglichen Gegenionen ab und nimmt eine äquivalente Menge Ionen aus der Lösung auf. Daneben kommt es zur Aufnahme von Lösungsmittel, was zur Quellung des Ionenaustauschers führt. Im GGW-Zustand liegen die beiden konkurrierenden Kationen in beiden Phasen (Lösung und Ionenaustauscher) entsprechend ihrer Affinitäten nebeneinander vor.
Der Ionenaustausch ist immer stöchiometrisch, elektroneutral und reversibel, was den entscheidenden Unterschied zur Adsorption ausmacht. Die Adsorption ist durch die Wechselwirkungen eines gelösten Stoffes mit einem festen Stoff vorwiegend durch physikalische Kräfte (van der Waals) charakterisiert. Oft liegen jedoch Adsorption und Ionenaustausch gemeinsam vor. Zur Charakterisierung dieses Austauschgleichgewichtes können verschiedene Kenngrößen verwendet werden. Die Lage des GGW einer Reaktion lässt sich durch formale Anwendung des Massenwirkungsgesetzes in Form der thermodynamischen GGW-Konstanten formulieren. Da Aktivitätskoeffizienten schwer zugänglich sind, verwendet man meist Selektivitätskoeffizienten. Der Selektivitätskoeffizient K beschreibt die Fähigkeit eines Ionenaustauschers, unter sonst gleichen Bedingungen, bestimmte Ionen gegenüber anderen bevorzugt aus der Lösung zu entfernen. Als Selektivität wird die auswählende Wirkung eines Austauschers bezeichnet, die auf den Aufbau des Ionenaustauschers im Allgemeinen und die Art und Konzentration aller Ionen in der Lösung zurückzuführen ist [Dorfner 1970]. Von großer Bedeutung für die Selektivität ist der Vernetzungsgrad des Polymers. Weiterhin spielen die Stärke der Wechselwirkungen zwischen Fest- und Gegenion eine Rolle. Liegen mehrere Ionen vor, so wird die Ionenart bevorzugt ausgetauscht, deren Selektivitätskoeffizient am größten ist. Im Gegensatz zur thermodynamischen Konstante ist der Selektivitätskoeffizient durch Konzentrationen oder Äquivalentanteile der Ionen in beiden Phasen definiert, d. h. er gilt nur für konstante äußere Bedingungen.
|
(Gl. 2.4) |
| ↓35 |
Bei chelatbildenden Austauschern ist zwischen Selektivität und Spezifität zu unterscheiden. Die bevorzugte Aufnahme bestimmter Ionen infolge der Spezifität beruht auf der chemischen Struktur der Ankergruppen, welche mit bestimmten Gegenionen besonders stabile Komplexe bilden. Die Bindungsenergien sind deshalb bei chelatbildenden Austauschern im Bereich von 60 - 100 kJ/mol, während sie bei gewöhnlichen Austauschern bei 8 – 12 kJ/mol liegen [Dorfner 1970].
Eine weitere wichtige Kenngröße ist die Verteilungsgewichtskonstante Kd in [mL/g], die durch das Konzentrationsverhältnis eines Ions zwischen Austauscher- und Lösungsphase im GGW beschrieben wird. Sie dient als Maß für die Verarmung oder Anreicherung eines Stoffes am Sorbenten.
Der Wert soll bei Anreicherung eines Ions möglichst hoch sein (Kd > 1000) [Knittel 1979].
| ↓36 |
|
(Gl. 2.5) |
Meist ist ein Abtrennen eines bestimmten Ions von anderen Ionen der Lösung das Ziel. Zur Ermittlung der Wirksamkeit des Ionenaustauschers wird dann der Trennfaktor α benutzt. Er ist als Quotient der Verteilungsverhältnisse definiert und unterscheidet sich vom Selektivitätskoeffizienten K dadurch, dass die Ionenwertigkeiten nicht exponentiell eingehen.
|
(Gl. 2.6) |
| ↓37 |
Um die Aufnahmefähigkeit (Kapazität) der einzelnen Ionenaustauscher darzustellen, trägt man die Beladung des Austauschers qeq gegen die Ionenkonzentration der Lösung im GGW ceq auf. So erhält man Sättigungskurven, deren Sättigungswerte der maximalen Beladung qmax. (Kapazität) der Ionenaustauscher entsprechen. Es wird davon ausgegangen, dass die aus der Lösung entfernte Menge stets der am Ionenaustauscher angereicherten Menge entspricht. Bei bekanntem Volumen V, unter der Annahme, dass das Volumen der flüssigen Phase konstant ist und definierter Anfangskonzentration c0, kann sie über die Massenbilanz hergeleitet und indirekt über die Bestimmung der GGW-Konzentration der flüssigen Phase ermittelt werden:
|
(Gl. 2.7) |
Unter der Voraussetzung, dass zu Beginn die unbeladene Probe q0 = 0 ist, ergibt sich für die GGW -Beladung qeq:
| ↓38 |
|
(Gl. 2.8) |
Parallel dazu kann die Beladung direkt aus den Ergebnissen der Eluate ermittelt werden. Zur grafischen Darstellung der Abhängigkeit der GGW-Beladung von der GGW-Konzentration sind Austauschisothermen geeignet, qeq = f(ceq bzw. peq)T. Es existieren verschiedene Modellvorstellungen zur mathematischen Beschreibung.
Die Anreicherung eines Stoffes an der Grenzfläche einer benachbarten Phase wird allgemein als Sorption bezeichnet. Findet die Anreicherung ausschließlich an der Oberfläche statt, so spricht man von Adsorption. Der Sorptionsprozess ist ein physikalisch-chemisches Trennverfahren, bei dem die zu sorbierende Stoffe (Adsorptiv) der Gas- oder Flüssigphase entsprechend Abb. 2.9.1 an der Oberfläche einer feste Phase (Adsorbens) angelagert werden. Dabei stellt sich ein thermodynamisches GGW zwischen Adsorpt (Adsorptiv im angelagerten Zustand) und Adsorptiv ein. Das Ausmaß der Adsorption eines Adsorptivs durch ein Adsorbens hängt von den Eigenschaften der beteiligten Substanzen, der Konzentration der Lösung und der Temperatur ab. Die Geschwindigkeit mit der sich das GGW einstellt wird durch mehrere Transportwiderstände bestimmt.
| ↓39 |
| Abb. 2.9.1: Prinzip der Adsorption [Kienzle, Bäder 1980] | ||
|
|
Die Beziehung zwischen der an der Oberfläche adsorbierten Menge und der freien Adsorptivkonzentration wird Adsorptionsisotherme genannt [Dörfler 1994]. Die Adsorptionsgeschwindigkeit ![]() ist proportional der Anzahl der unbesetzten aktiven Stellen (qmax – qi) und der Konzentration ci:
ist proportional der Anzahl der unbesetzten aktiven Stellen (qmax – qi) und der Konzentration ci:
|
(Gl. 2.9) |
| ↓40 |
Die Desorptionsgeschwindigkeit ![]() ist proportional der Anzahl der besetzten aktiven Plätze qi:
ist proportional der Anzahl der besetzten aktiven Plätze qi:
|
(Gl. 2.10) |
Es gibt eine Vielzahl verschiedener Modellvorstellungen zur mathematischen Beschreibung der Adsorption, die entweder empirischer Natur sind oder ihren Ursprung in theoretischen Modellvorstellungen haben. Darüber hinaus unterscheiden sich die Ansätze hinsichtlich der Anwendbarkeit auf Einstoff- oder Mehrstoffgemische [Eltner 1998]. Von zahlreichen in der Literatur erwähnten Modellen, sind die bekanntesten die von Langmuir und Freundlich. Diese haben sich in der Praxis bewährt und beschreiben den Verlauf der Adsorptionsisothermen mit guter Genauigkeit.
| ↓41 |
Zur mathematischen Beschreibung der Austauschisothermen verdient die von Langmuir aufgestellte Gleichung für eine Adsorptionsisotherme besondere Aufmerksamkeit, da hier zahlreiche experimentelle Werte in erster Näherung richtig beschrieben werden können. Das von Langmuir entwickelte Sorptionsmodell wurde ursprünglich für die Adsorption von Gasen an festen Oberflächen hergeleitet und geht von folgenden vier Annahmen aus [Langmuir 1918]:
im GGW sind Adsorptions- und Desorptionsgeschwindigkeit gleich (![]() =
=![]() ):
):
| ↓42 |
|
(Gl. 2.11) |
Nach Langmuir wird mit steigender Adsorptivkonzentration eine maximale Beladung der Adsorbensoberflächen erreicht. Die Parameter qmax und K bezeichnen die maximale Beladungskapazität des Adsorbens bei Annahme einer monomolekularen Bedeckung (qmax) und die Affinität von Adsorbens und Adsorptiv (K) unter den gegebenen Versuchsbedingungen [Bathen, Breitbach 2001] [Wilke 2001]. Die mathematische Formulierung der Abhängigkeit von der GGW-Beladung und –konzentration lautet:
|
(Gl. 2.12) |
| ↓43 |
Die Ermittlung der systemabhängigen Konstanten qmax und K9 erfolgt durch grafische Auswertung der Messdaten der linearisierten Form (Gl. 2.13).
|
(Gl. 2.13) |
So können die Langmuir-Parameter aus der Steigung (1/qmax) und aus dem Ordinatenabschnitt (1/K∙qmax) bei ceq = 0 ermittelt werden. In einigen Veröffentlichungen wird die Langmuir-Beziehung in anderer Form unter Verwendung der Langmuir-Konstante b genutzt. Beide Gleichungen sind durch
b = 1/Kbzw. b = kdes/ka ineinander überführbar [Kley 1999]. Vorteil der ausgewählten Modellvorstellung nach Langmuir ist die Tatsache, dass in der Berechnung eine maximale Beladung der monomolekularen Bedeckung der Oberfläche berücksichtigt wird und die Abschätzung für weite Konzentrationsbereiche gültig ist.
| ↓44 |
| Abb. 2.9.2: Isothermendarstellung nach Modellen von Langmuir und Freundlich [Wilke 2001] | ||
|
|
Obwohl das Langmuir-Modell auf einer definierten Anzahl von Bindungsplätzen des Adsorbens basiert und häufig Überlagerungen unterschiedlicher Bindungsmechanismen vorliegen (Adsorption, Ionenaustausch, Komplexierung, Chelatbildung), beschreibt das Modell diesen komplexen Vorgang meist mit hoher Genauigkeit. Im Grenzfall sehr kleiner Konzentrationen geht die Langmuir-Isotherme in eine Lineare über (Henry-Bereich), hier ist die Beladung linear abhängig von der Konzentration ceq. Im Bereich hoher Konzentrationen steigt sie langsamer an bis sie einen Sättigungswert erreicht. Die Isotherme zeigt dann eine waagerechte Isothermenform (Abb. 2.9.2). Je mehr energetisch günstige Stellen besetzt sind, desto geringer wird die Wahrscheinlichkeit, dass eine weitere Adsorption stattfindet. Nach Absättigung aller verfügbaren Bindungsstellen des Adsorbens lässt sich die Beladung nicht weiter steigern. Die Beladung hängt nicht mehr von der GGW-Konzentration der Lösung ab. Man spricht dann von einer horizontalen oder indifferenten Isothermengleichung [Sontheimer et al. 1985].
Die Langmuir-Isotherme geht von einer monomolekularen Bedeckung aus, die in der Praxis jedoch selten auftritt. Das von Freundlich empirisch entwickelte Modell (Gl. 2.14) enthält eine Potenzfunktion zur Beschreibung der Isothermen, darin ist kF die Freundlich-Konstante und nF der Freundlich Exponent. Das Freundlich-Modell geht von der Annahme aus, dass die Adsorptionsenergie nicht gleich groß ist. Zu Beginn der Beladung, wo noch viele freie Plätze vorhanden sind, ist die Adsorptionsenergie hoch. Je mehr Plätze bereits besetzt sind, umso kleiner wird sie.
| ↓45 |
|
(Gl. 2.14) |
Durch Logarithmieren kann die allgemeine Freundlich-Gleichung (Gl. 2.14) in eine Geradengleichung umgeformt werden (Gl. 2.15).
|
(Gl. 2.15) |
| ↓46 |
Im Gegensatz zum Langmuir-Modell ergibt sich bei hohen GGW-Konzentrationen keine maximale Beladung. Durch grafische Auswertung können die Freundlich-Parameter nF und kF bestimmt werden.
Neben den bisher vorgestellten Isothermen, die eine monomolekulare Adsorption beschreiben, entwickelten Brunauer, Emmett und Teller die BET-Theorie [Rouquerol et al. 1999]. Diese berücksichtigt die Möglichkeit der Mehrfachbedeckung, wobei für jede einzelne Schicht eine ideal lokalisierte Monoschichtadsorption nach Langmuir vorausgesetzt wird.
Für die Ausbildung mehrerer Adsorbatschichten und den Übergang zur Kapillarkondensation sind van-der-Waals-Kräfte verantwortlich, die durch hohe Gasdrücke und tiefe Temperaturen begünstigt werden. Bei der BET-Isotherme erfolgt keine Sättigung. In ihrem Verlauf bis zum Wendepunkt entspricht sie der Langmuir-Isotherme. Molekularer Stickstoff kann unter den Bedingungen niedriger Temperaturen und niedriger Drücke mit den meisten Feststoffen nicht chemisch reagieren. Er lagert sich reversibel, in Abhängigkeit vom vorherrschenden Gasdruck, an der Feststoffoberfläche an. Deshalb wird Stickstoff zur Bestimmung der spezifischen Oberfläche und der Verteilung der Porengrößen verwendet.
| ↓47 |
Als Adsorptions-Isotherme ist die adsorbierte Gasmenge na (z.B. N2) als Ordinate in mol/g gegen den jeweiligen Relativdruck p/p0 als Abzisse aufgetragen (p0 = 101,32 kPa, Sättigungsdampfdruck des N2 bei 77 K) [Klank 2005].
Die Auswertung nach BET basiert auf der ISO 9277 (DIN 66131). Sie wird zur Berechnung der Adsorbat-Monoschichtkapazität und der spezifischen Oberfläche bei der Stickstoffsorption angewandt. Die Monoschichtkapazität n m ist aus der BET-Gleichung (2.16) zu berechnen.
|
(Gl. 2.16) |
| ↓48 |
Durch lineare Darstellung der BET-Isothermen (Abb. 2.9.3, Gl. 2.16) im Bereich der Relativdrücke 0,05 ≤ p/p0 ≤ 0,3lassen sich der BET-Kennwert C sowie die Gasmenge einer monomolekularen Gasschicht n m bestimmen.
| Abb. 2.9.3: BET-Diagramm [ISO 9277] | ||
|
|
Die Steigung b und der Ordinatenschnittpunkt a lassen sich grafisch oder durch lineare Regression ermitteln. Daraus errechnen sich die Monoschichtkapazität nm und der BET-Kennwert C (Gl. 2.17).
| ↓49 |
|
(Gl. 2.17) |
Die auf die entgaste Probenmasse bezogene spezifische Oberfläche as in m2∙g-1 berechnet sich nach Gleichung 2.18 aus der Monoschichtkapazität nm multipliziert mit der Avogadrokonstante L(6,022∙1023) und dem Flächenbedarf eines Adsorbatmoleküls am (am für N2 = 0,162 nm2).
|
(Gl. 2.18) |
4 Besser Hydratationsradius der Ionen
5 [Nightingal 1959], [Appel, Ma 2002]
6 * Keine Angaben in der Literatur verfügbar
7 Im verzerrten Oktaeder sind die axialen Liganden schwächer gebunden als die äquatorialen und weisen demnach einen größeren Bindungsabstand auf.
8 Im September 2004 wurden die Polymeraktivitäten auf die Lanxess Deutschland GmbH übertragen
9 Definitionsgemäß ist die Konstante K = ka/kdes
| © Die inhaltliche Zusammenstellung und Aufmachung dieser Publikation sowie die elektronische Verarbeitung sind urheberrechtlich geschützt. Jede Verwertung, die nicht ausdrücklich vom Urheberrechtsgesetz zugelassen ist, bedarf der vorherigen Zustimmung. Das gilt insbesondere für die Vervielfältigung, die Bearbeitung und Einspeicherung und Verarbeitung in elektronische Systeme. | ||
| DiML DTD Version 4.0 | Publikationsserver der Universität Potsdam | HTML-Version erstellt am: 21.05.2007 |